.png)
Main page

24 - 29 September 2023
The goal of the workshop is to introduce you to several computational chemistry concepts and allow you to interactively explore them through Jupyter Notebooks.
You will need...
| To bring your own laptop... | To have prior knowledge of Quantum Mechanics... | To pre-install eChem a few days before the workshop (instructions here). |
 |
 |
 |
Topics
-
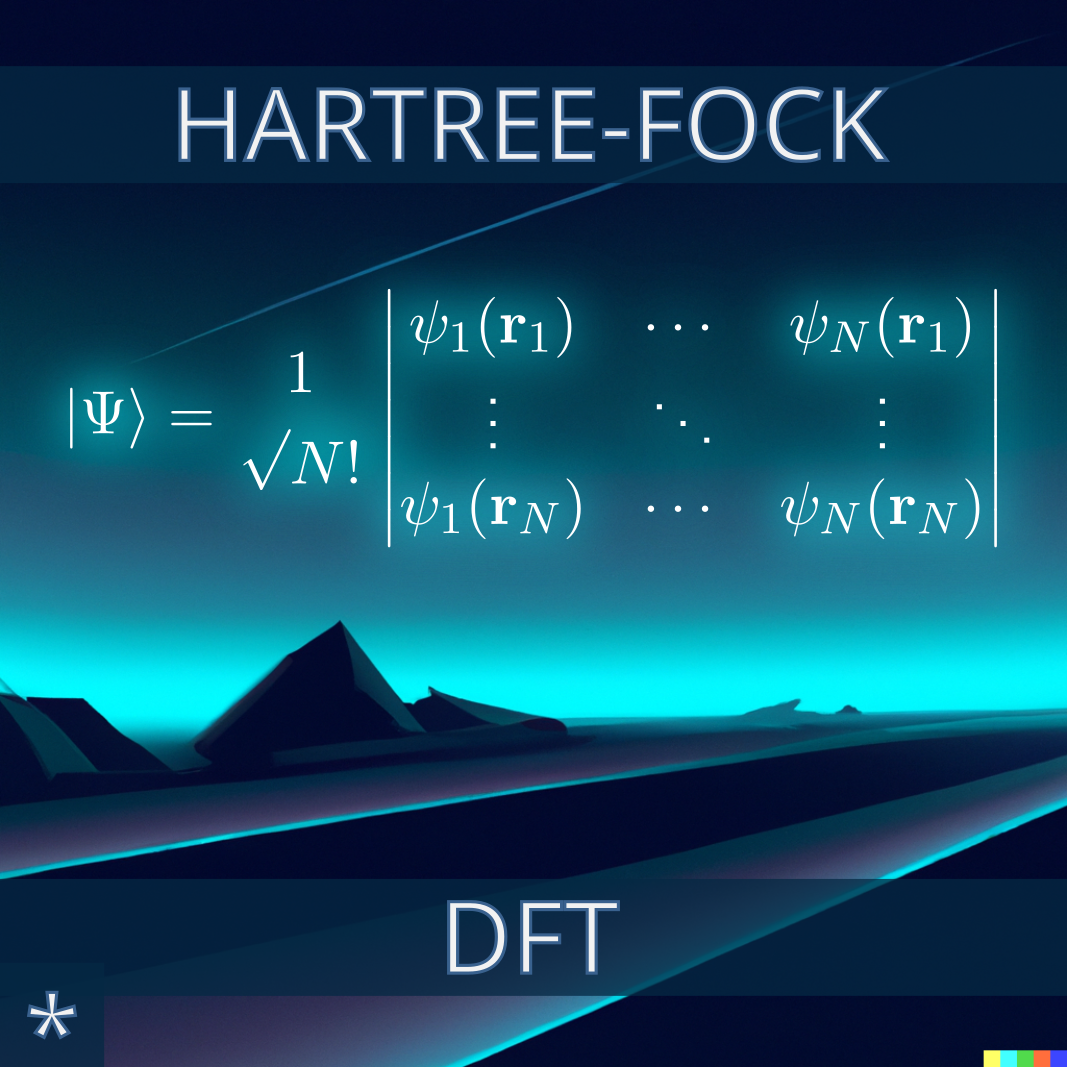
Hartree–Fock (HF) Theory
-
We will write our own HF program for SCF energy minimizations, use it to determine molecular properties like the dipole moment and explore its potential in a Quantum Mechanics / Molecular Mechanics setup.
-
Density Functional Theory (DFT)
-
Starting from the HF routines, we will write our own Kohn-Sham DFT program and use it to explore DFT-specific issues such as the self-interaction error.
-
Electron Correlation
-
We will study of a weakly-bonded dimer and analyze why going beyond HF is necessary in order to correctly describe such a system.
-
Structure Optimization
-
We will write our own algorithms to optimize molecular structures and compare their performance in terms of computing time and number of optimization steps.
-
Molecular Dynamics
-
We will parametrize our own force field for a small molecule and use it to perform classical dynamics simulations in gas-phase and in solution.
Week Overview
| Sunday | Monday | Tuesday |
 |
 |
 |
| Wednesday | Thursday | Friday |
 |
 |
 |
Let us know if you will be attending by 15 June 1 July 2023!
There are 30 spots available and the registration fee is 500 PLN or 110 EUR. The registration feee will cover the coffee breaks and social events. The Center of Excellence Astrochemistry and Astrophysics will fund a limited number of grants for accommodation in shared rooms at the University Hotel. Please indicate your interest in the accommodation grant in the registration form. Payment instructions and account number will be sent in a separate e-mail after the confirmed registration.
Register (closed)
(Note: you do not need to submit an abstract)
*The pictures marked with and asterisk contain backgrounds or elements created with the help of DALLE-2